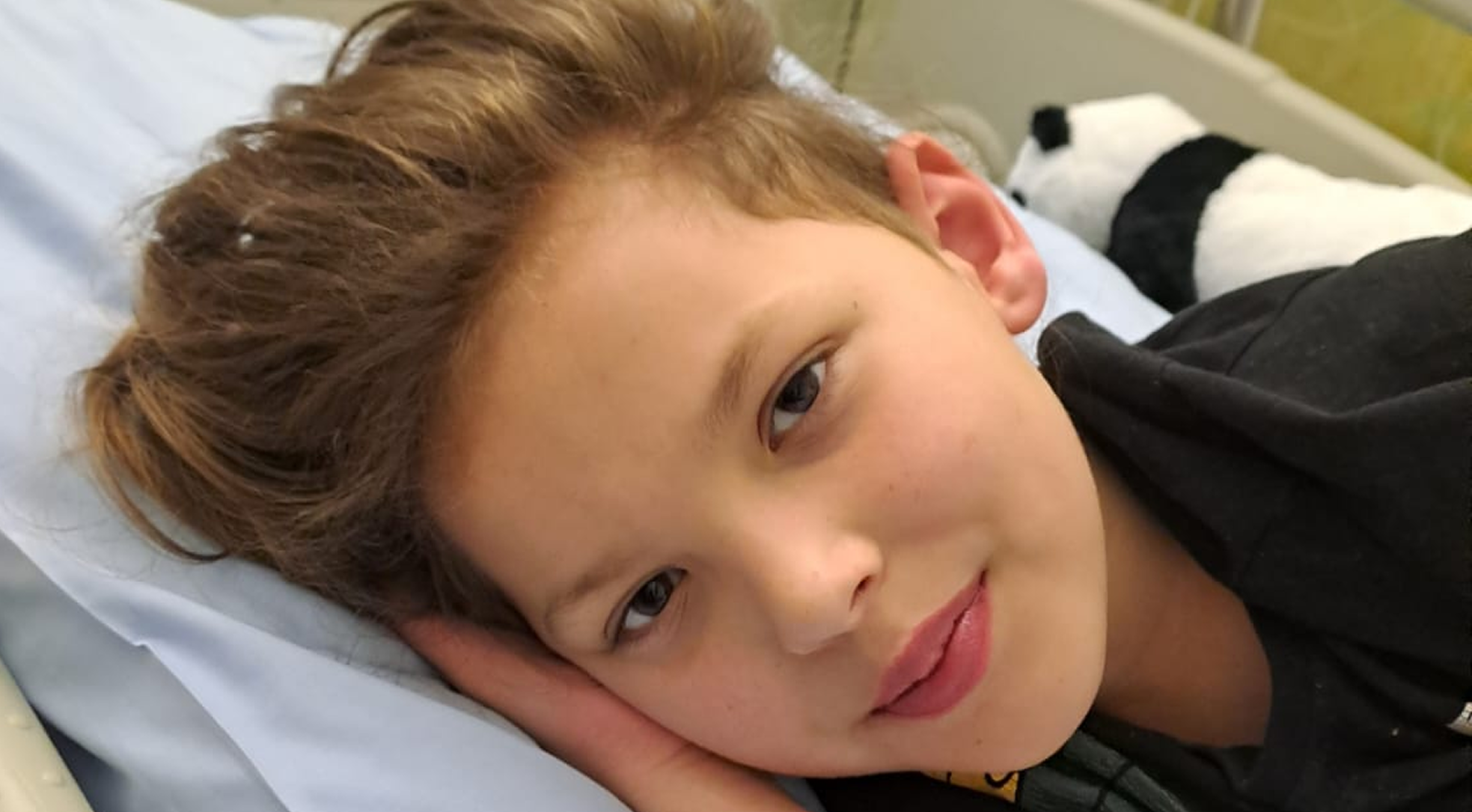
Maksym Duhanets
Maksym ma 8 lat. To bystry, wrażliwy chłopiec, który z pasją rysuje, buduje z klocków LEGO i...






Choroba nowotworowa to dla dzieci i nastolatków ogromne wyzwanie. Dzięki Twojej pomocy i naszemu wsparciu nasi Podopieczni mogą mu sprostać.
Wpłać datek Przekaż 1,5%


Każda darowizna ma znaczenie. Chcesz pomóc? Wpłać dowolną kwotę na Fundację i jej Podopiecznych.
Dowiedz się więcejWsparcie oferowane przez biznes ma ogromne znaczenie dla naszych Podopiecznych, a firmom pozwala zbudować wizerunek.
Dowiedz się więcejChcesz pomóc bezpośrednio? Rozwijaj swój potencjał i dołącz do społeczności wolontariuszy Fundacji.
Dowiedz się więcej

Rozwój jest podstawą współczesnej medycyny. Dlatego wsparcie badań i innowacji jest dla Fundacji kluczowym elementem działania. Dzięki temu pomagamy w tworzeniu nowych narzędzi do walki z chorobami nowotworowymi.
Jednym z naszych głównych celów jest budowanie świadomości w społeczeństwie oraz edukacja dzieci i ich opiekunów, a także wolontariuszy, lekarzy i personelu medycznego przygotowujących się do niesienia pomocy najmłodszym.
Osoby po udanej terapii wciąż mogą wymagać pomocy. Potrzebują wiedzy, jak zadbać o siebie po chorobie, często wymagają dopasowanej opieki oraz wsparcia psychologicznego. Dostrzegamy potrzeby Ozdrowieńców i wspieramy ich.
Nowotwory to nie tylko choroba ciała. Doświadczenie nowotworu ma wpływ na psychikę i emocje pacjentów oraz ich bliskich. Dlatego wszechstronna pomoc zapewniana przez Fundację ma tak duże znaczenie.
Rozwój jest podstawą współczesnej medycyny. Dlatego wsparcie badań i innowacji jest dla Fundacji kluczowym elementem działania. Dzięki temu pomagamy w tworzeniu nowych narzędzi do walki z chorobami nowotworowymi.
Jednym z naszych głównych celów jest budowanie świadomości w społeczeństwie oraz edukacja dzieci i ich opiekunów, a także wolontariuszy, lekarzy i personelu medycznego przygotowujących się do niesienia pomocy najmłodszym.
Osoby po udanej terapii wciąż mogą wymagać pomocy. Potrzebują wiedzy, jak zadbać o siebie po chorobie, często wymagają dopasowanej opieki oraz wsparcia psychologicznego. Dostrzegamy potrzeby Ozdrowieńców i wspieramy ich.
Nowotwory to nie tylko choroba ciała. Doświadczenie nowotworu ma wpływ na psychikę i emocje pacjentów oraz ich bliskich. Dlatego wszechstronna pomoc zapewniana przez Fundację ma tak duże znaczenie.

Maksym ma 8 lat. To bystry, wrażliwy chłopiec, który z pasją rysuje, buduje z klocków LEGO i...

Nadia ma sześć lat i jest prawdziwym wulkanem energii. Trudno za nią nadążyć – ciągle w ruchu,...