
ALL jest to ponowne pojawienie się choroby po okresie remisji. Więcej niż 85% dzieci z ostra białaczką limfoblastyczną (ALL) ma przeżycie wolne od wznowy po konwencjonalnej terapii, natomiast przeżycie po wznowie choroby jest dużo gorsze. Wznowa ALL, wynikająca z lekooporności komórek nowotworowych, stanowi istotny problem kliniczny.

Czym jest nawrót ALL?
Jest to ponowne pojawienie się choroby po okresie remisji. Więcej niż 85% dzieci z ostra białaczką limfoblastyczną (ALL) ma przeżycie wolne od wznowy po konwencjonalnej terapii, natomiast przeżycie po wznowie choroby jest dużo gorsze. Wznowa ALL, wynikająca z lekooporności komórek nowotworowych, stanowi istotny problem kliniczny. Metody postępowania diagnostycznego przyjęte przez Polskie Towarzystwo Hematologii i Onkologii Dziecięcej zostały, opracowane na podstawie protokołu leczenia pierwszego nawrotu ALL u dzieci: IntReALL HR 2010 wersja 2.0 z 10.12.2015 (dla grupy wysokiego ryzyka) oraz IntReALL SR 2010 wersja 1.91 z 12.08.2016 przez Międzynarodową Grupę Roboczą ds. Lekoopornej Białaczki u Dziecii (Resistant Disease Committee of the International BFM Study Group).
Gdzie umiejscawia się nawrót ALL?
Nawrót białaczki może wystąpić w szpiku (wznowa szpikowa) jak i w narządach pozaszpikowych (wznowa pozaszpikowa) – najczęściej w ośrodkowym układzie nerwowym (OUN), dodatkowo u chłopców w jądrach, poza tym możliwa jest lokalizacja w każdym narządzie. W zależności od miejsca nacieków komórek białaczkowych definiuje się wznowę jako izolowaną (szpikową lub pozaszpikową) albo mieszaną (szpikową i pozaszpikową), co zostało przedstawione w tabeli:

W przypadku podejrzenia nawrotu szpikowego zajęcie szpiku kostnego ocenia się za pomocą mikroskopu świetlnego z zastosowaniem kryteriów francusko-amerykańsko-brytyjskich (FAB). Wymagane są aspiracje szpiku kostnego z dwóch stron (zwykle obu tylnych kolców biodrowych). Jeśli aspiracja jest niewystarczająca (sucha punkcja), należy wykonać trepanobiopsję szpiku kostnego.
W przypadku nawrotu w ośrodkowym układzie nerwowym, o nawrocie świadczy obecność limfoblastów w płynie mózgowo-rdzeniowym (PMR) i pleocytoza powyżej 5/ul komórek białaczkowych (status CNS3). Jeśli ilość blastów jest <5/ul, diagnozowany jest status CNS2, który nie jest klasyfikowany jako zajęcie OUN. Jeśli PMR jest zanieczyszczony krwią i wykrywa się w nim blasty, to rozpoznaje się izolowany nawrót w OUN w przypadku, gdy we krwi obwodowej nie stwierdza się obecności limfoblastów. W przypadku obecności blastów w krwi obwodowej stosuje się algorytm Steinherz/Bleyer: jeśli stosunek WBC/RBC do liczby limfoblastów w PMR przekracza stosunek w krwi obwodowej >2 razy, zakłada się zajęcie OUN. W przeciwnym razie podejrzewa się zanieczyszczenie. W sytuacjach niejasnych może być konieczna indywidualna decyzja. Jeśli blasty zostaną wykryte w PMR bez spełnienia kryteriów zajęcia OUN (CNS2), pacjent otrzymuje zintensyfikowaną chemioterapię dooponową podobnie jak pacjenci z zajęciem OUN. Nie następuje jednak dodatkowe napromienianie OUN. Obecność nawrotu OUN powinna być również potwierdzona za pomocą dostępnych metod diagnostycznych (TK głowy, MRI mózgowia), nawet w przypadku braku pleocytozy w PMR, kiedy występują kliniczne objawy zajęcia OUN, takie jak zaburzenia widzenia, polifagia, porażenia. Jeżeli naciekanie opon mózgowo-rdzeniowych stwierdza się tylko w badaniach obrazowych, a wznowa białaczki nie jest jednoznacznie udowodniona (brak innych lokalizacji nacieków białaczkowych), może być konieczne wykonanie biopsji.
W przypadku nawrotu jądrowego o zajęciu jąder świadczy jedno- lub obustronne bezbolesne powiększenie jąder z naciekiem limfoblastów potwierdzonym biopsją. Zakres powiększenia musi być udokumentowany z użyciem orchidometru lub badaniem USG moszny. W przypadku klinicznie prawidłowego lub niejednoznacznego jądra kontralateralnego należy wykonać biopsję w celu wykluczenia lub potwierdzenia subklinicznego zajęcia jadra.
W przypadku innych lokalizacji nawrotu pozaszpikowego każde miejsce, narząd lub tkanka może być infiltrowane przez komórki białaczkowe. Aby wykryć klinicznie niejawne zajęcie narządów, należy wykonać badanie ultrasonograficzne brzucha, miednicy i regionalnych węzłów chłonnych oraz RTG klatki piersiowej. Dodatkowo inne badania obrazowe, takie jak MRI lub TK, mogą być konieczne do rozpoznania zajęcia narządów pozaszpikowych.
Jak przebiega diagnostyka nawrotu ALL?
Diagnoza nawrotu ALL oparta jest na cytologicznych, immunologicznych, cytogenetycznych i molekularnych badaniach genetycznych wykorzystywanych do optymalnej stratyfikacji leczenia oraz oceny jego skuteczności. Kompleksowa charakterystyka komórek białaczkowych, zwłaszcza z użyciem badań molekularnych, stwarza możliwość identyfikacji markerów, które mogą być potencjalnym celem w terapii biologicznej.
Badanie cytologiczne rozmazów szpiku kostnego, krwi obwodowej oraz PMR wykonuje się, stosując klasyczne barwienie May’a-Grünwalda-Giemsy, które pozwala na identyfikację morfologicznie nieprawidłowych, niedojrzałych komórek w różnych proporcjach. Charakterystyka morfologiczna na podstawie systemu klasyfikacji FAB może być wykorzystana do identyfikacji linii komórek białaczkowych.
Immunofenotypowanie z użyciem wielokolorowego cytometru przepływowego jest podstawową metodą umożliwiającą identyfikację komórek białaczkowych na podstawie oceny markerów powierzchniowych i cytoplazmatycznych. Konieczny do ustalenia grup ryzyka podział komórek na prekursory limfocytów B (BCP) lub T jest przeprowadzany zgodnie z klasyfikacją EGIL. W nawrocie BCP-ALL szczególnie istotna jest ocena ekspresji antygenów, takich jak CD19 i CD22 jako punktów docelowych dla terapii biologicznej. Immunofenotypowanie przeprowadza się w aspiracie szpiku kostnego w momencie rozpoznania. Immunofenotypowanie PMR u pacjentów z nawrotem OUN nie jest obowiązkowe, ale zalecane.
Identyfikacja markerów do monitorowania minimalnej choroby resztkowej (MRD) polega naIlościowej ocenie MRD na poziomie genomu za pomocą łańcuchowej reakcji polimerazy (PCR) z zastosowaniem rearanżacji klonalnego genu receptora komórek T (TCR)/immunoglobuliny (IG) i jest uważana za złoty standard. Równolegle z badaniem PCR konieczna jest wyjściowa ocena specyficznych dla klonu białaczkowego markerów immunologicznych z użyciem wielokolorowej cytometrii przepływowej. Dostarczają one ważnych informacji o ekspresji antygenu w resztkowych komórkach białaczkowych podczas różnych faz leczenia, a także danych o regeneracji szpiku w czasie terapii. Jeśli materiał diagnostyczny jest skąpy, oznaczanie MRD metodą PCR jest metodą preferowaną. Zaleca się identyfikację co najmniej dwóch markerów do oznaczenia ilościowego MRD metodą PCR w szpiku kostnym. Jeśli dostępny jest tylko jeden marker, wyniki są konfrontowane z wynikami cytometrii przepływowej. Jeśli wyniki obu metod są spójne, są one akceptowane do stratyfikacji. Natomiast jeśli są sprzeczne, MRD tego punktu czasowego może być uznane jako „niewykonane/niedostępne”. Wszystkie zalecenia dotyczące metod oznaczania specyficznych markerów do monitorowania MRD są zgodne z wytycznymi grupy Euro-MRD (dawna nazwa: European Study Group on MRD detection in ALL).
Informacje o strukturalnych i numerycznych zmianach chromosomów są istotne do kompleksowej biologicznej charakterystyki nawrotu ALL. W związku z tym ocena powszechnych genów fuzyjnych, stanu ploidii i kariotypu cytogenetycznego są zawarte w rutynowych analizach genetyczno-molekularnych dla wszystkich grup ryzyka. Ocenę genów fuzyjnych, stanu ploidii i kariotypu przeprowadza się w aspiratach szpiku kostnego w momencie rozpoznania.
Powszechne geny fuzyjne raportowane w dziecięcej ALL, takie jak ETV6-RUNX1, BCR-ABL, E2A-PBX1, MLL-AF4 i MLL-ENL, są sprawdzane w momencie nawrotu w celu potwierdzenia pojawienia się ponownie wcześniej zidentyfikowanego genu fuzyjnego lub identyfikacji nowego. Zazwyczaj stosuje się metody: molekularną identyfikację genetyczną transkryptu genu fuzyjnego metodą PCR i analizę cytogenetyczną za pomocą metody fluorescencyjnej.
Ocena stanu ploidii w celu zidentyfikowania hipo- lub hiperdiploidalności jest przeprowadzana z użyciem kariotypu, metodą FISH z sondą centromerową i innymi sondami lub chromosomem systemu Multiprobe-I oraz w wyniku pomiaru indeksu DNA. Ocenę indeksu DNA przeprowadza się przy użyciu jodku propidyny do barwienia DNA mierzonego metodą cytometrii przepływowej. Aby zidentyfikować nadmiar lub utratę chromosomów, konieczne jest oszacowanie klasycznego kariotypu lub zastosowanie nowszych metod genetyki molekularnej, takich jak multipleksowa zależna od ligacji amplifikacja sond lub polimorfizm pojedynczych nukleotydów/porównawczych macierzy hybrydyzacji genomowej.
Cytogenetyczny kariotyp limfoblastów może być użyty do identyfikacji anomalii chromosomowych, takich jak aneuploidia, translokacje chromosomalne z wykluczeniem ukrytej translokacji i delecji. Komórki mitotyczne są poddawane obróbce chemicznej w celu uzyskania pasma G (Giemsa) lub Q (chinakryna). Konwencjonalne kariotypowanie jest metodą uzyskania globalnej analizy genomowej.
Jakie są czynniki ryzyka, grupy ryzyka w nawrocie ALL u dzieci i z czym to się wiąże?
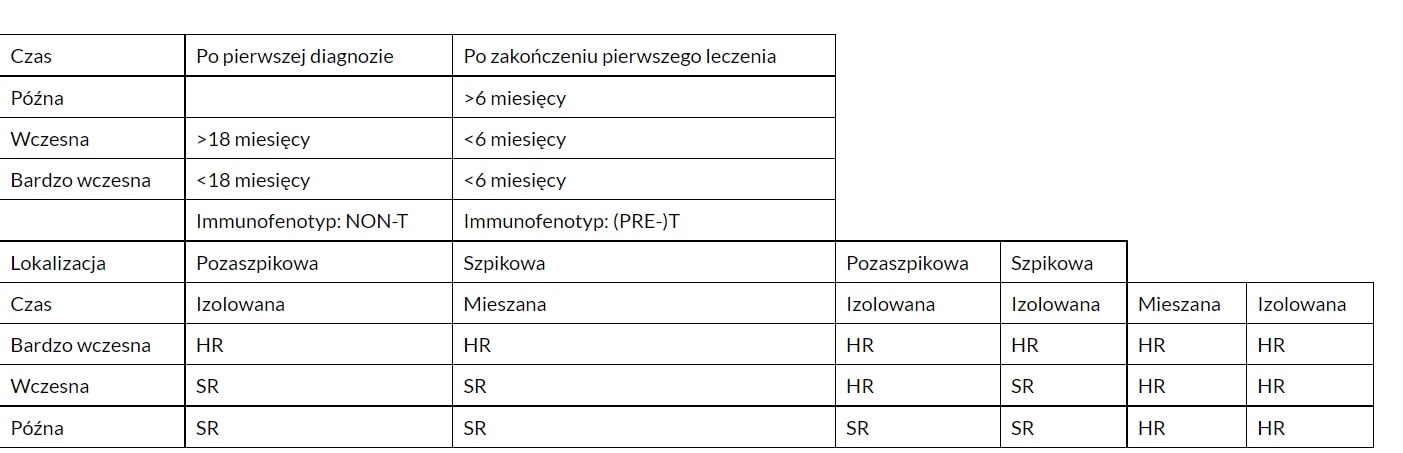
Czynniki ryzyka pierwszego nawrotu ALL mające wpływ na przeżycie zostały zawarte w schemacie stratyfikacji: Czas od diagnozy do wznowy (krótszy gorzej rokuje), miejsce wznowy (wznowa szpikowa gorsza niż pozaszpikowa), immunofenotyp (T gorszy niż B) oraz minimalna choroba resztkowa (MRD) w odpowiedzi na reindukcje. Po ustaleniu rozpoznania nawrotu ALL przed rozpoczęciem terapii należy zakwalifikować pacjenta do grupy ryzyka standardowego (SR) lub wysokiego (HR). Kwalifikację do grupy ryzyka w zależności od czasu wystąpienia wznowy, lokalizacji oraz immunofenotypu przedstawiono w tabeli:
Jak leczony jest pierwszy nawrót ALL u dzieci?
Leczenie pierwszego nawrotu ALL w grupie standardowego ryzyka (SR) prowadzone jest według protokołu IntReALL 2010 SR (realizowanego od 2013 r.), a w grupie wysokiego ryzyka (HR) według protokołu IntReALL 2010 HR (realizowanego po modyfikacji od 2019 r.).
Protokół IntReALL 2010 SR zawiera fazy: indukcji remisji, konsolidacji, leczenia podtrzymującego remisję oraz postępowania zapobiegającego rozwojowi białaczki OUN. W grupie SR stosowana jest terapia według ramienia SR-A (oparta na stosowanym wcześniej protokole niemieckim ALL-REZ BFM 2002) lub SR-B (oparta na protokole brytyjskim UK ALLR3). Po zakończeniu cyklu indukcyjnego oceniana jest remisja (mielogram i MRD zalecaną metodą RQ-PCR), co ma kluczowe znaczenie przy wyborze opcji terapeutycznej na późniejszym etapie leczenia. U pacjentów z późną mieszana lub późną szpikową wznową, u których po cyklu indukcyjnym stwierdza się remisję (w szpiku < 5% komórek blastycznych, brak ognisk pozaszpikowych) i niski poziom MRD (w 28 dniu < 10^-3 dla ramienia SRA lub w 35 dniu < 10^-4 dla ramienia SRB) oraz u pacjentów z izolowaną późną wznową pozaszpikową (jeśli osiągnęli remisję) stosuje się chemioterapię: leczenie indukcyjne, konsolidujące, podtrzymujące remisję oraz lokalną radioterapię. U wszystkich pozostałych dzieci należy przeprowadzić procedurę wysoko dawkowanej radioterapii i chemioterapii z transplantacją allogenicznych komórek hematopoetycznych (allo-HSCT) w 14-17 tygodniu leczenia.
Protokół IntReALL 2010 HR zawiera fazy: indukcji remisji (chemioterapia według zmodyfikowanego protokołu UK ALL R3 z mitoksantronem) oraz konsolidacji (zmodyfikowany protokół AIEOP-BFM 2009). W fazie leczenia indukcyjnego stosuje sie 2 schematy terapii (randomizacja lub wybór): ramię A – chemioterapia lub chemioterapia z dodatkiem bortezomibu (inhibitora proteasomu 268). Wykazano działanie synergiczne bortezomibu z dopuszczalną toksycznością w połączeniu z deksametazonem, doksorubicyną, winkrystyną i PEG-asparaginazą u dzieci z oporną na leczenie lub nawrotową ALL. Dlatego też spodziewany jest wiekszy efekt cytotoksyczny i wyższy niż dotychczas odsetek uzyskanych remisji. W grupie HR wszyscy pacjenci, którzy osiągną remisję hematologiczną, powinni być poddani transplantacji hematopoetycznych komórek macierzystych w 14 tygodniu po 4 cyklach chemioterapii.
A co w przypadku braku remisji, progresji lub kolejnego nawrotu ALL u dziecka?
Strategie leczenia są różne w lekoopornej nawrotowej ALL. Obecnie nie ma jednolitej opcji terapii w przypadku lekoopornej wznowy ALL u dzieci – decyzje podejmowane są indywidualnie. W przypadku nieuzyskania remisji hematologicznej lub przetrwania MRD przed planowaną transplantacją (> 10^-3), a także w razie wystąpienia kolejnej wznowy/progresji należy podjąć leczenie z użyciem cytostatyków niestosowanych wcześniej lub leków biologicznych. W chemioterapii lekoopornej ALL u dzieci zarejestrowane zostały 2 cytostatyki: klofarabina (antymetabolit nukleozydu puryn) – dla ALL linii B oraz nelarabina (anty-metabolit nukleozydu puryn) – dla ALL linii T. Najczęściej stosuje się je w kombinacji z cyklofosfamidem i vepesidem jako terapię pomostową przed procedurą transplantacji macierzystych komórek krwiotwórczych. Obecnie prowadzone są badania różnych leków biologicznych, jednak nieliczne były dotychczas stosowane w lekoopornej ALL u dzieci. Spośród nich tylko inhibitor proteasomu – bortezomib (stosowany obecnie w ramieniu B cyklu indukcyjnego IntReALL 2010 HR) w skojarzeniu z chemioterapią okazał się efektywny zarówno w ALL linii B, jak i linii T (badania nierandomizowane). Najlepiej udowodnione zostało działanie blinatumomabu – przeciwciała dwuspecyficznego CD19/CD3, które kojarzy blasty CD19+ z limfocytami T pacjenta, dzięki czemu możliwe jest uruchomienie mechanizmów immunologicznych prowadzących do destrukcji komórki białaczkowej. Dotychczasowe wyniki badan klinicznych przeprowadzone w lekoopornej ALL CD19+ u dzieci wykazały wysoką skuteczność (ok. 90% remisji molekularnej) oraz akceptowalną toksyczność (głównie zespół uwalniania cytokin oraz powikłania neurologiczne) mniejszą niż agresywna chemioterapia. Blinatumomab został zaakceptowany jako optymalna opcja terapii pomostowej przed transplantacją. Inotuzumab ozogamycyny – przeciwciało anty-CD22 sprzężone z kalicheamycyną, która ma działanie cytotoksyczne, może być stosowany w monoterapii lub w skojarzeniu z chemioterapią. Lek jest zarejestrowany w leczeniu lekoopornej ALL CD22+ u dorosłych. W związku z działaniem hepatotoksycznym kalicheamycyny u pacjentów poddanych transplantacji zwiększa się ryzyko choroby okluzyjnej naczyń wątroby (VOD/SOS). Modyfikowane genetycznie, aktywowane limfocyty CAR-T stanowią obiecującą opcję terapii, jednak dotychczas możliwe jest zastosowanie ich tylko w przypadku lekoopornej ALL CD19+. Obserwacja pacjentów poddanych tej terapii jest zbyt krótka, aby można było zdecydować, czy jest to terapia ostateczna, czy też, jak w przypadku innych stosowanych leków stanowi leczenie pomostowe przed allo-HSCT.
Rekomenduje się także przy rozpoznaniu nawrotu ALL wykonanie szerokiego badania molekularnego limfoblastów i ustalenie celów do indywidualnej terapii biologicznej z udziałem leków eksperymentalnych.
Czy są jakieś nowości terapeutyczne na horyzoncie?
W aktualnie opracowywanym protokole leczenia pierwszego nawrotu ALL przewiduje się zastosowanie inotuzumabu w leczeniu indukcyjnym, i/lub blinatumomabu w skojarzeniu z chemioterapią w leczeniu poindukcyjnym. Dla części pacjentów z grupy bardzo wysokiego ryzyka w leczeniu poindukcyjnym rozważa się terapię CAR-T, która być może zakończy leczenie bez następczej transplantacji. Nowy protokół leczenia pierwszego nawrotu ALL u dzieci jest jeszcze we wczesnej fazie przygotowania, wobec czego koncepcje mogą ulec jeszcze zmianie w zależności od wyników toczących się badań klinicznych.
Ewa Gorczyńska, Monika Mielcarek-Siedziuk, Joanna Owoc-Lempach



